
—— Estudo colaborativo do Centro de Controle e Prevenção de Doenças de Zhejiang, Macro & Micro-Test e Centro de Controle e Prevenção de Doenças da China publicado na revista Frontiers in Cellular and Infection Microbiology
Visão geral do estudo
Em maio de 2026, a revista Frontiers in Cellular and Infection Microbiology (JCR Q1, IF ≈ 4,6) publicou um artigo liderado pelo Centro Provincial de Controle e Prevenção de Doenças de Zhejiang (Zhejiang CDC), com a equipe de bioinformática da Beijing Macro & Micro-Test Bio-Tech Co., Ltd. e o Instituto Nacional de Controle e Prevenção de Doenças Transmissíveis (China CDC) como coautores. O estudo tem o seguinte título:
“Identificação e análise filogenética de sete cepas de Brucella abortus em Zhejiang, China.”

Este estudo representa a primeira análise sistemática de rastreabilidade filogenética baseada em genoma completo de Brucella abortus (B. abortus) na província de Zhejiang, China. A equipe analisou sete isolados coletados entre 2015 e 2025 (quatro cepas de origem humana e três de origem bovina de Jinhua, Quzhou e Ningbo). Os resultados fornecem evidências genômicas sobre a origem e as rotas de transmissão dessa “espécie dominante no norte” em uma região epidêmica atípica no sul do leste da China.
Contexto e Significado
A brucelose é uma zoonose causada por bactérias do gênero Brucella. A Brucella abortus infecta principalmente o gado, mas também pode causar doenças em humanos. Na China, a brucelose apresenta variações geográficas significativas: a maior incidência ocorre nas províncias do norte (por exemplo, Mongólia Interior, Shanxi, Heilongjiang). Em contrapartida, as províncias do sul, incluindo Zhejiang, têm sido historicamente dominadas pela Brucella melitensis, com poucos casos relatados de B. abortus. Essa disparidade regional torna a caracterização genética e o rastreamento da origem da B. abortus em Zhejiang uma prioridade fundamental de saúde pública.
Métodos e principais resultados
A equipe de pesquisa adotou uma estratégia multifacetada que combina biologia molecular e bioinformática:
1.Identificação de patógenos e tipagem básica
Os testes de PCR para o gene BCSP-31 e AMOS-PCR confirmaram que todos os sete isolados eram B. abortus.
A tipagem de sequência multilocus (MLST) baseada em nove genes de manutenção revelou que todos os isolados pertenciam ao tipo de sequência ST2, indicando alta homogeneidade genética entre as cepas circulantes de B. abortus em Zhejiang.
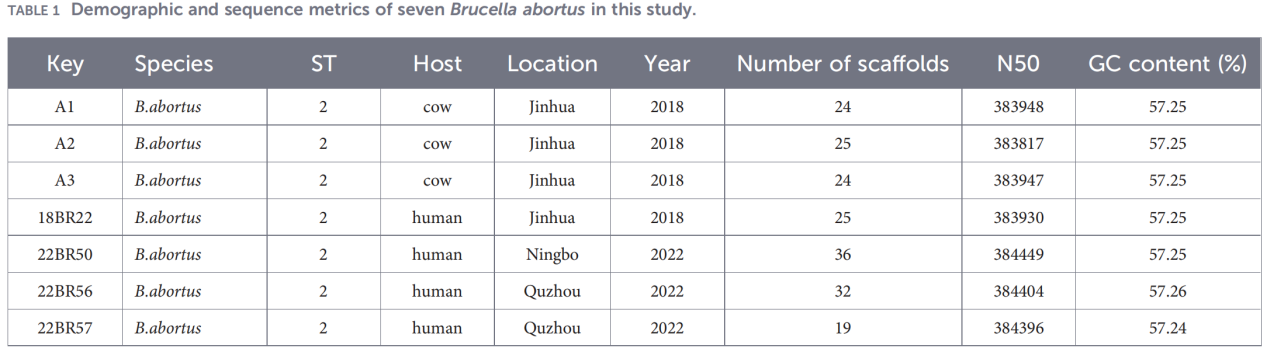
2.Caracterização do genoma completo
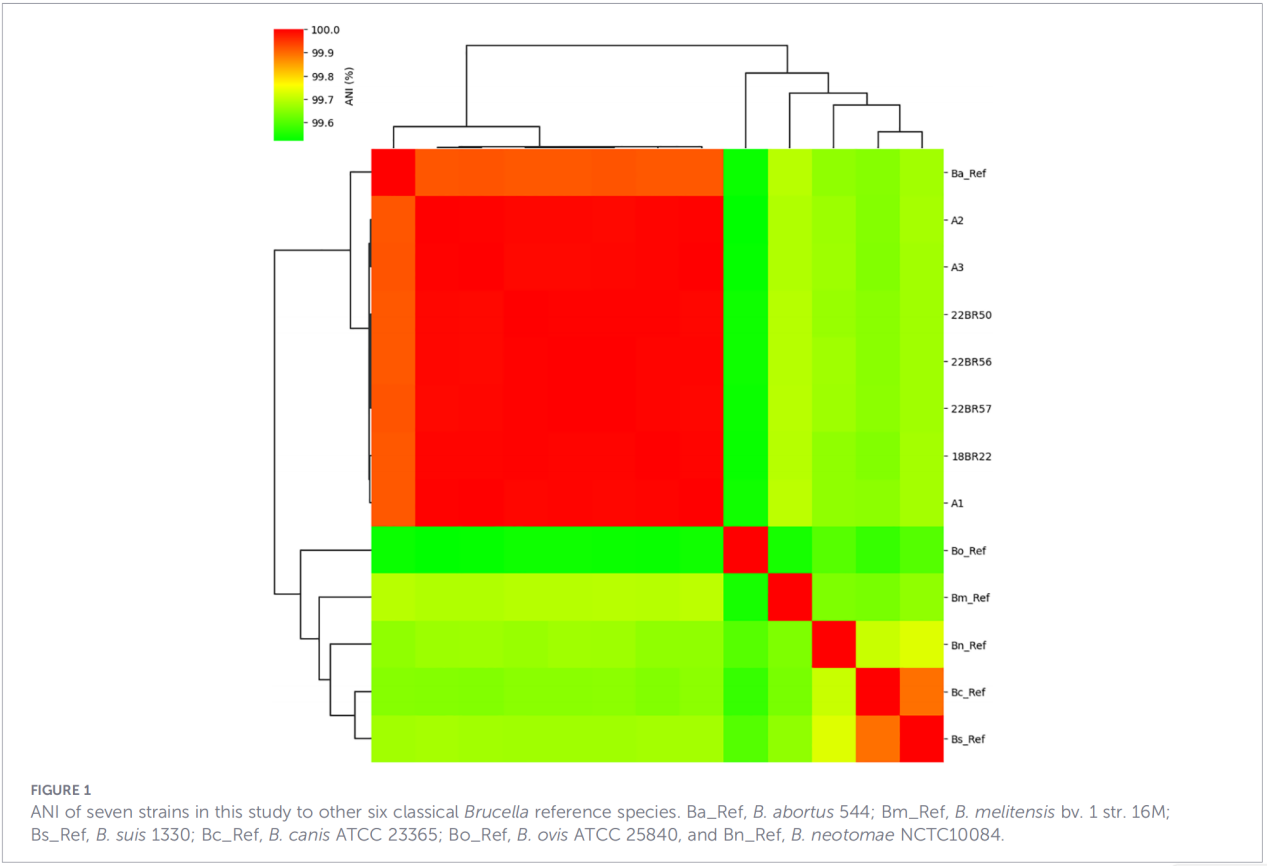
O sequenciamento do genoma completo foi realizado na plataforma Illumina NovaSeq. A análise de identidade nucleotídica média (ANI) mostrou que os isolados de Zhejiang compartilhavam até 99,99% de similaridade com a cepa de referência B. abortus 544.
A análise do pan-genoma revelou uma população altamente conservada: foram identificados 3.084 genes essenciais, juntamente com apenas 10 genes da casca, e nenhum gene de núcleo mole ou de nuvem foi detectado.
3.Perfis de genes de virulência e resistência antimicrobiana
Um total de 68 fatores relacionados à virulência foram previstos, abrangendo vias clássicas como a biossíntese de LPS, o sistema de secreção T4SS e o sistema regulatório de dois componentes BvrR-BvrS. Notavelmente, todos os isolados não apresentaram os genes de adesina bmaA e btaF. A análise de genes de resistência detectou apenas o gene mprF no banco de dados CARD, sem que outros determinantes de resistência fossem identificados.
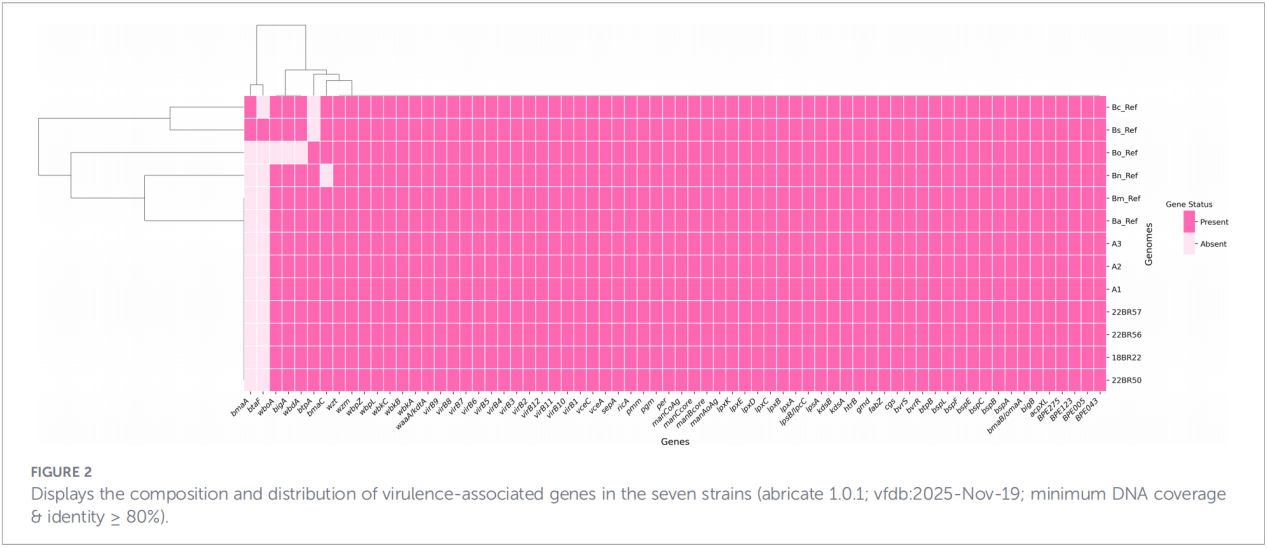 4. Reconstrução filogenética e rastreamento da transmissão
4. Reconstrução filogenética e rastreamento da transmissão
A análise de polimorfismos de nucleotídeo único do genoma central (cgSNP) posicionou os isolados de Zhejiang em uma posição específica na árvore filogenética global. Os resultados mostraram que as cepas de Zhejiang formam um grupo monofilético juntamente com cepas da Rússia, Mongólia e várias províncias do norte da China (Ningxia, Heilongjiang, Mongólia Interior, Hebei, Gansu, Pequim). Este grupo se subdivide em três subclados distintos (Clado 1–3), sugerindo múltiplos eventos independentes de introdução.
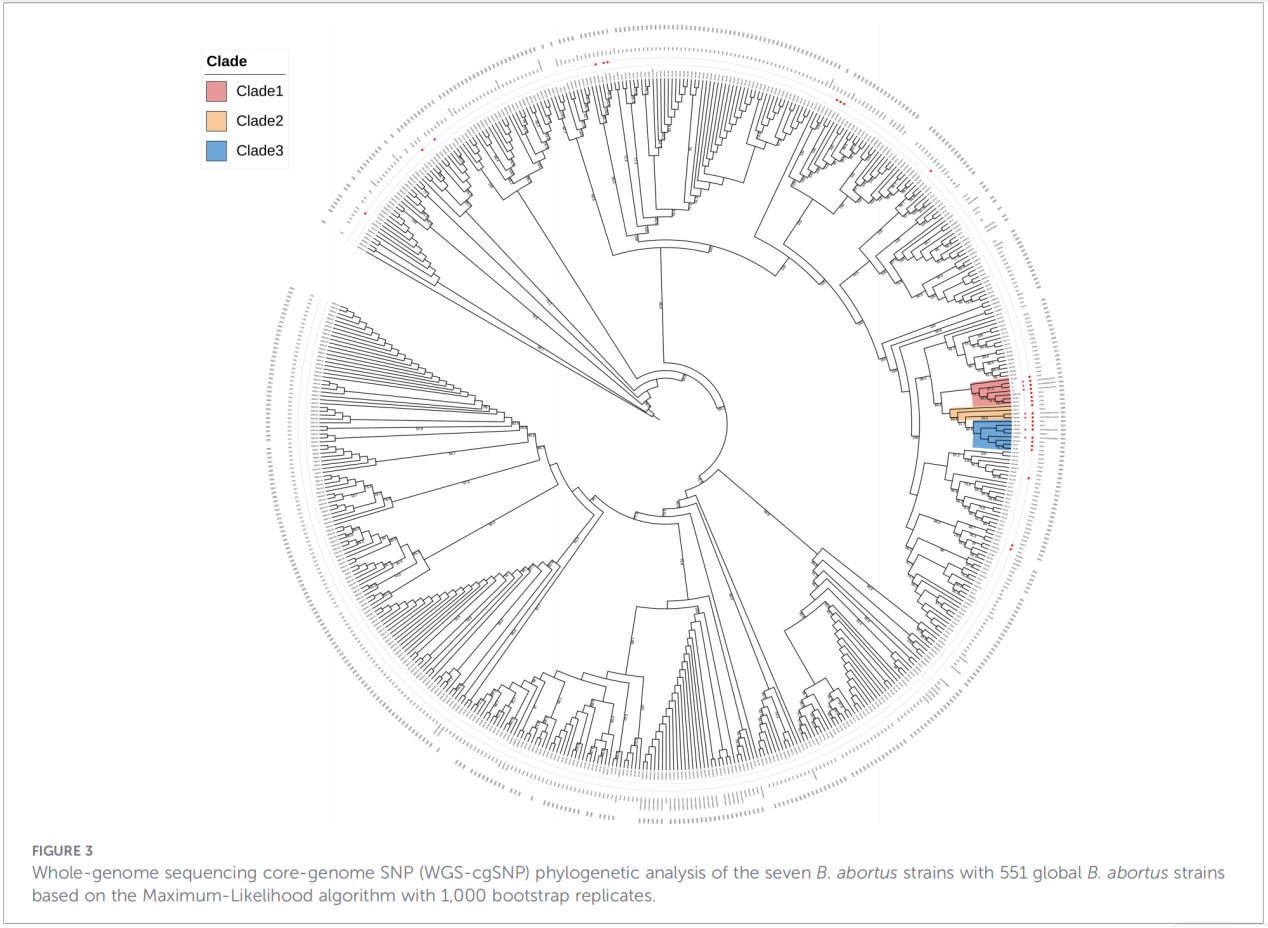
Conclusões e implicações
Este estudo fornece o primeiro conjunto de dados genômicos de alta precisão de B. abortus na província de Zhejiang e apresenta diversas conclusões importantes:
- Clear fundo genético– As cepas de B. abortus que circulam em Zhejiang pertencem ao ST2, são altamente conservadas genomicamente e representam uma linhagem típica de brucelose bovina.
2. Evidensidade de transmissão inter-regional– A análise filogenética não corrobora a existência de uma linhagem endêmica independente em Zhejiang. Em vez disso, os dados sugerem fortemente que essas cepas se originaram no norte da China e podem compartilhar uma base evolutiva comum com cepas da Rússia e da Mongólia. A presença de três subclados implica múltiplos eventos de introdução distintos.
3. Implicações para a saúde pública– Os resultados reforçam a importância da vigilância genômica da brucelose, mesmo em regiões tradicionalmente não endêmicas, como Zhejiang. Embora o número atual de casos seja baixo, ferramentas de alta resolução como o cgSNP podem rastrear eficazmente a origem de surtos importados e fornecer evidências científicas para interromper as cadeias de transmissão associadas ao transporte interprovincial de animais.
Este trabalho não só preenche uma lacuna de pesquisa na província de Zhejiang, como também fornece novos dados de referência para a vigilância de patógenos e a avaliação de risco da brucelose na região do Delta do Rio Yangtzé.
Informações sobre o artigo:
Yang, Y., Shi, X., Chen, J., Wang, L., Wu, Z., Yao, W., … & Wu, B. (2026). Identificação e análise filogenética de sete cepas de Brucella abortus em Zhejiang, China. Frontiers in Cellular and Infection Microbiology, 16, 1758965.
Data da publicação: 10 de junho de 2026